
Industry: Medical Devices
 The Challenge
The Challenge
Addressing FDA requirements for Software Tool Validation of Off-The-Shelf (OTS) Software in previously developed Tool Qualification Kits.
 Background
Background
Genuen (formerly CertTech LLC) had previously worked with a leading software vendor of Test and Requirements Management Software Tools to develop Software Tool Qualification Kits for their software. Their software was used in product development and product manufacturing quality process steps and Software Tool Qualification of such tools was required by the vendor’s clients working in the Automotive and Functional Safety Industries.
In response to those needs, Genuen developed the reference documentation and procedures required by the clients to demonstrate the suitability of the vendor’s software for developing and manufacturing their products in accordance with the applicable safety standards (IEC 61508; ISO 26262)
After completion of the original Software Tool Qualification kits, Genuen was approached by the Software Vendor with a request to support the Software Validation of the same tools for use in the medical industry. This involved performing a ‘gap analysis’ between the content of the Software Tool Qualification Kits used by end-users in the Functional Safety and Automotive industries and the requirements to be met to address the Software Validation of Software Tools used in the Medical industry.
 The Solution
The Solution
Software Qualification vs Medical Software Validation: Preliminary Gap Analysis
The vendor’s clients in the Medical industry cited the following two sections of the Code of Federal Regulations (CFR) that were driving their tool validation needs:
-
Title 21 Part 820.70(i) : Covering the Quality System Regulations on Production and Process Controls (regulated by the FDA), and
-
Title 21 Part 11 : Covering Electronic Records and Electronic Signatures (for submittals to the FDA)
Title 21 Part 820.70(i)
The key wording in this CFR driving Software Validation is “..When computers or automated data processing systems are used as part of the production or quality system, the manufacturer shall validate computer software for its intended use according to an established protocol..”.
Note that the CFR does not say HOW to validate the computer software, but the absence of such details is similar to that found in the standards written for the Functional Safety, Automotive, Rail, Aerospace, and other industries where high reliability and highly safe software is essential. While the CFR does not promote specific methodologies for validating applicable software, we found that the FDA does recognize several ‘consensus standards’ that pertain to software validation. The following two were considered to be the most applicable:
-
AAMI TIR 36: Validation of Software for Regulated Processes, and
-
General Principles of Software Validation (GPSV)
A determination if there were any gaps in our original Software Tool Qualification kits necessitated that we compare the validation methods described in these two documents against the qualification methods that had been originally employed for the Software Tools.Title 21 CFR Part 11: Our reading of this regulation was that its focus was not directly related to achieving confidence in Software Tools used in the Medical Industry. Instead, it was primarily aimed at providing regulatory guidance on the objectives (i.e. requirements) to be satisfied by Software Tools software that produced electronic records needed for submittal to the FDA and on the requirements for signatures stored in electronic form vs those recorded on paper submittals.
With respect to 21 CFR Part 11, a gap existed in the original Software Tool Qualification Kits for the vendor’s tools since they did not perform any evaluation of the Software Tools as to whether they met the objectives of this regulation. To address the needs of the Medical industry would require adding the necessary documentation and tests to these Qualification Kits.
Comparison of the Medical Industry Software Validation Methods vs Software Tool Qualification
From a high-level view – the Software Tool Qualification methods that were already being performed to meet the standards of the Functional Safety and Automotive Industry could be correlated with those specified in the FDA’s consensus standards. Each industry segment may have used different terminology but essentially described the following common objectives to be met as part of Software Validation/Qualification:
-
Describe WHAT the USER needs it to do.
-
OBJECTIVELY show that the Software performs those tasks CORRECTLY.
-
DOCUMENT HOW to repeatably perform the validation tasks to support later regression testing upon installation of vendor supplied software updates or user customizations of the software.
The first bullet focuses on what the User expects from the Software and requires that the user record those expectations in the form of a High Level Use Cases (denoting the major functional capabilities that the user relies upon from the Software) followed by more detailed software requirements against which the objective verification testing can be performed. Commercial Software Tool vendors do not generally provide the detailed requirements, but the User defined High Level Use Cases can typically be informed from the vendor supplied User Manuals.
Before proceeding, it should be made clear that Software Validation activities are not limited to simply ‘testing’ a software application against its requirements. For custom software, or software developed in-house, the validation methods used to establish confidence in the software tool include code reviews, white-box software testing, integration testing, among others. However, for commercial off-the-shelf software the options available to an end-user are typically limited to testing unless software vendor is very cooperative and is willing to share their internal records pertaining to the verification activities they performed during development and prior to shipment. End users might take into consideration the vendor’s reputation, the length of time the software has been in service, and the record of software patches the vendor has had to perform to address residual defects, but these are only ‘subjective’ metrics and can only be used to supplement objective software quality evidence collected by the users themselves.
One of the consensus standards accepted by the FDA, TIR 36, addressed using Risk-Based metrics for determining how much rigor is required during the validation activities. Per TIR 36, the level of validation effort was determined by the severity of anticipated harm in the event an error existed in the tool plus the probability of the presence of an error. If the level of harm was low, or the probability of an error was low in a given software feature then less validation effort would be required for that feature than if the potential harm was high or the probability was high. This risk-based approach for driving validation rigor had already been integrated in the original Tool Qualification packages. A simple example being that the Editing Features (those used to write the Requirements or Test Scripts) did not include validation procedures because errors in the editing feature would be immediately apparent to the user and/or reviewers. All such decisions regarding which features to validate (or not) were documented in the Safety Manual(s) provided in the tool qualification kits (a partial example is provided below).
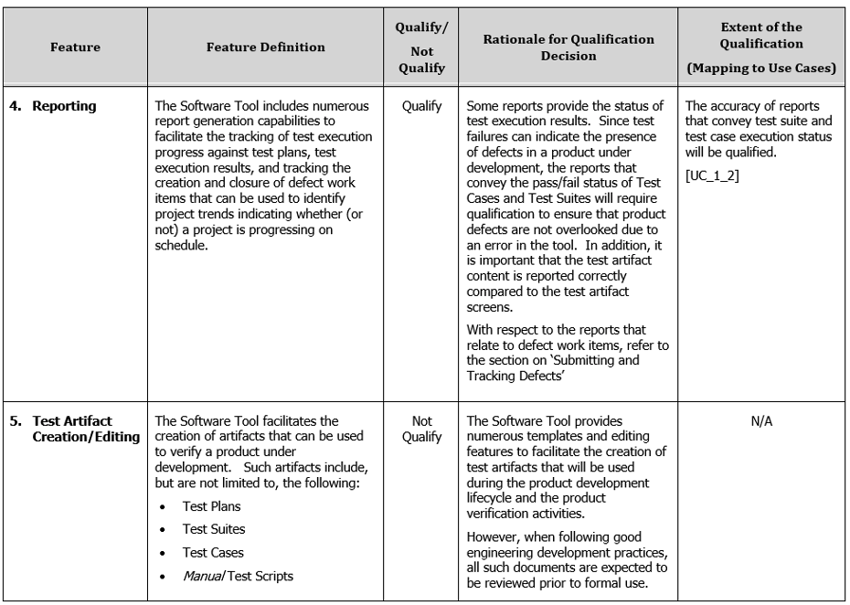
Note that one other ‘gate’ determining whether a feature is requires validation involves whether that feature is actually used. If not, it would be perfectly valid to identify such cases in the Safety Manual and defer validation of those features if it later became necessary. However, in our case, we treated all Software Features as if they would potentially be used.
The following documentation addressed the needs of the Medical Industry with only minor updates:
Safety Manual
This identified WHAT features required validation based on a risk-based assessment
Requirements Manual
This identified in the detailed requirements for each Software Use-Case identified in the Safety Manual as requiring Validation
Trace Matrix
This cross referenced the Detailed Requirements and Use-Cases to specific tests (source code or manual test procedures
Validation Methods (whether automated or manual)
These documented methods provided the repeatable validation ‘protocol’ referenced in Title 21 Part 820.70(i) and the results of executing these methods provided objective evidence that the Software satisfied the user-defined software requirements.
Addressing Title 21 Part 11
These required adding requirements and validation methods to the Software Tool Qualification Kits to cover Medical Industry specific regulations. To ensure each of the objectives of the regulation were addressed a mapping table was included in the Safety Manual as shown in the partial example below:
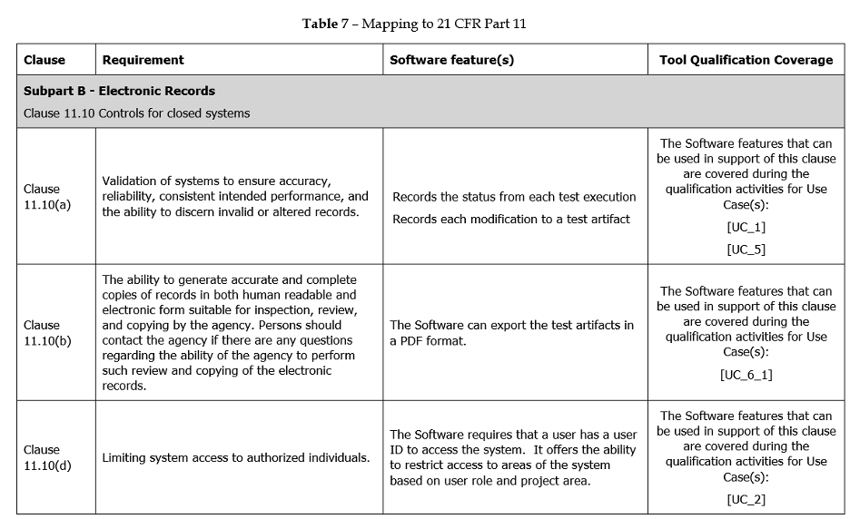
Summary
Genuen has extensive experience is assisting clients in qualifying their software tools in many industries – of which Medical is only one.
Ready to Get Started?
Learn more about our products or request a consultation with an experienced engineer.
